Porcine pleuropneumonia is a contagious disease caused by Actinobacillus pleuropneumoniae (App) [1-3]. App is a Gram-negative bacterium that can be differentiated into 18 serotypes [4]. Several virulence factors are involved in the pathogenesis of App, including capsule polysaccharide, lipopolysaccharide, proteases, transferrin-binding proteins, outer membrane proteins, and Apx toxins [5]. All serotypes of App can variously produce four different Apx toxins, namely ApxI, ApxII, ApxIII, and ApxIV, which have potential value for use in vaccines and diagnostic tests. At least four genes, apxC, apxA, apxB, and apxD, are required for activated Apx toxins to be released from the bacterial cells [1,2]. The apxC gene encodes a protein that activates the preApx toxins by post-translational acylation. The apxA gene encodes the toxin structure. The apxB and apxD genes encode exporters that are capable of secreting Apx toxins [1,2].
Strains of App serotype 2 retain the apxIBD, apxIICA, and apxIIICABD genes [1,6]. Many ApxII-producing App serotypes such as serotype 2 can release this toxin via the ApxIB/ApxID exporter [1,7]. With the exception of serotype 3, all serotypes that do not produce ApxI possess a truncated apxI operon consisting of apxIB and apxID and are capable of producing the ApxIB/ApxID exporter that functions in ApxII secretion. However, the function of the ApxIB/ApxID exporter in the secretion of ApxIII is relatively under-studied in those serotypes. In this study, we constructed a mutant strain of App serotype 2 (1536; American Type Culture Collection, USA) by inactivation of apxIB and apxID to analyze the specificities of the ApxIB/ApxID exporter to the ApxIIA and ApxIIIA toxins.
A pBKS-App-Kanr recombination vector was designed to contain a fragment of consecutively arranged apxIB and apxID genes (GenBank accession number X68595.1) that were inactivated by the introduction of a Kanr gene (Fig. 1A). For the plasmid preparation, a 4.0 kb-long fragment of the apxIB and apxID genes was amplified from 1536 genomic DNA by polymerase chain reaction (PCR) using the primers apxIBD-F and apxIBD-R (Table 1). The PCR product was cloned into pTOP TA V2 (Enzynomics, Korea) to produce pTOP-App. pTOP-App was digested with SpeI and XhoI, and the 4.1 kb insert was ligated into pBluescript II KS(+) (Stratagene, USA) to obtain pBKS-App. Escherichia coli DH5╬▒ was used for plasmid DNA amplification. pBKS-App was digested with BamHI and MfeI to delete a 748 bp fragment of the apxIB and apxID genes. A fragment encoding Kanr was amplified from pECFP-C1 (BD Biosciences, USA) using the primers KanR-F and KanR-R. The PCR product was inserted into the BamHI and MfeI digested pBKS-App to form pBKS-App-Kanr.
Transformation of the 1536 strain was performed with pBKS-App-Kanr plasmid DNA linearized by SpeI. The App strains were cultured with PPLO-based media as described previously [8]. A transformant (ΔIBD) was selected by PCR screening with three primer pairs, SP1/apxID-B, SP1/apxID-A, and apxIB-A/SP2 (Fig. 1B, 1C, and 1D). In order to confirm the recombination, the PCR amplicons for primer pairs SP1/apxID-B and apxIB-A/SP2 were sequenced with primers SP2 and SP1, respectively (data not shown).
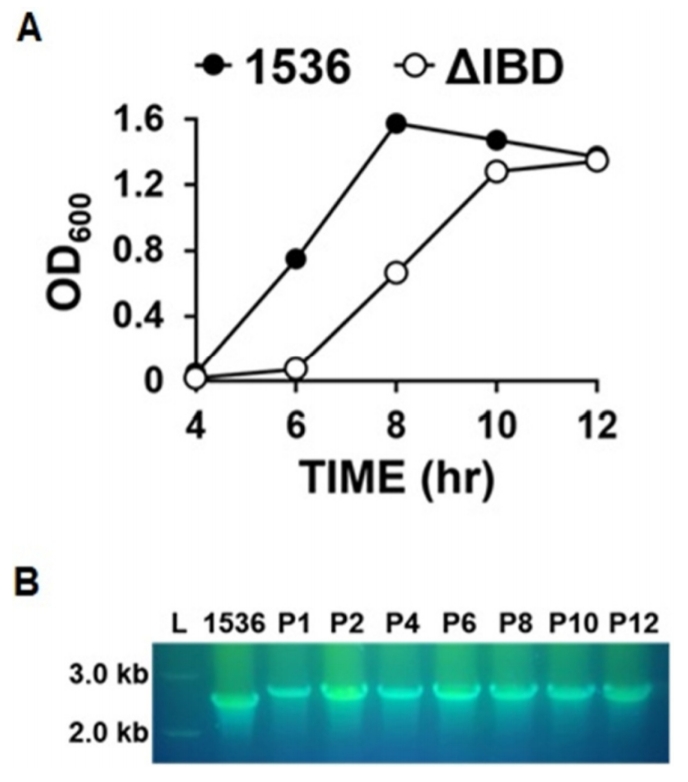
To obtain the growth curves of ΔIBD, overnight cultures were diluted at 1:1,000 and grown for 12 h. The OD600 of the culture was determined at intervals of 2 h with the Ultrospec 2000 UV/visible spectrophotometer (Pharmacia, Stockholm, Sweden). An obvious difference in the growth rate was observed between the ΔIBD and 1536 strains (Fig. 2A). ΔIBD showed a delayed growth rate compared to 1536, suggesting that the inactivation of the apxIB and apxID genes has a negative influence on the viability of App. The decreased growth rate might be due to cellular stress induced by unreleased Apx toxins. Despite the reduced growth rate, ΔIBD was found to be genetically stable during 12 successive passages in culture (Fig. 2B).
To express a fusion recombinant protein named Apx132, a 1,345 bp gene fragment was synthesized containing parts of ApxI (amino acids 460-578; GenBank accession number WP_005598583.1), ApxIII (amino acids 643-798; AAK 50053.1), and ApxII (amino acids 834-956; AAU84700.1). Fifteen-residue linkers coding for (GGGGS)3 were added between the ApxI and ApxIII parts and between the ApxIII and ApxII parts. The gene fragment was inserted into the linearized pET28a(+) (Novagen, UK), and the recombinant plasmid was then transformed into E. coli BL21 (DE3). Selected colonies were cultured at 37┬░C in LB media supplemented with 100 ╬╝g/mL kanamycin. Overnight cultures (diluted 1:100) were grown until mid-log phase at which point protein expression was induced for 4 h by the addition of isopropyl-╬▓-D-thiogalactopyranoside to a final concentration of 1 mM. The cells were washed twice with ice-cold PBS (pH 7.4) and then lysed for 30 min in Buffer A (100 mM sodium phosphate, 10 mM Tris-HCl, 100 ╬╝g/mL lysozyme and 5 mM phenylmethanesulfonylfluoride (PMSF), pH 8.0). The lysate was centrifuged at 10,000├Śg for 20 min, and the resulting pellet was resuspended by incubation for 1 h at room temperature in Buffer B (8 M urea, 10 mM Tris-HCl, 100 mM sodium phosphate and 5 mM PMSF; pH 8.0). The solubilized extracts were then loaded onto a Ni-nitrilotriacetic acid column (Qiagen, USA). The column was washed with Buffer C (8 M urea, 10 mM Tris-HCl, 100 mM sodium phosphate and 5 mM PMSF; pH 6.3) and eluted with Buffers D and E (8 M urea, 10 mM Tris-HCl, 100 mM sodium phosphate and 5 mM PMSF; pH 5.9 and 4.5, respectively).
Apx132 protein was purified with a Ni-nitrilotriacetic acid column (Qiagen) and used for the preparation of an antiApx132 antiserum in mice. Female Balb/c mice aged 6 to 8 weeks (SLC Japan, Japan) were housed in laboratory animal facilities at the College of Veterinary Medicine, Seoul National University. The mice were immunized by subcutaneous administration of 25 ╬╝g of immunogen on Days 0, 14, 28, and 42. Immunogens were administered in complete FreundŌĆÖs adjuvant on Day 0 and in incomplete FreundŌĆÖs adjuvant on Days 14, 28, and 42. Serum samples were obtained by retro-orbital bleeding under anesthesia on Day 63. Western blots were performed with anti-Apx132 antiserum and horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G secondary antibodies.
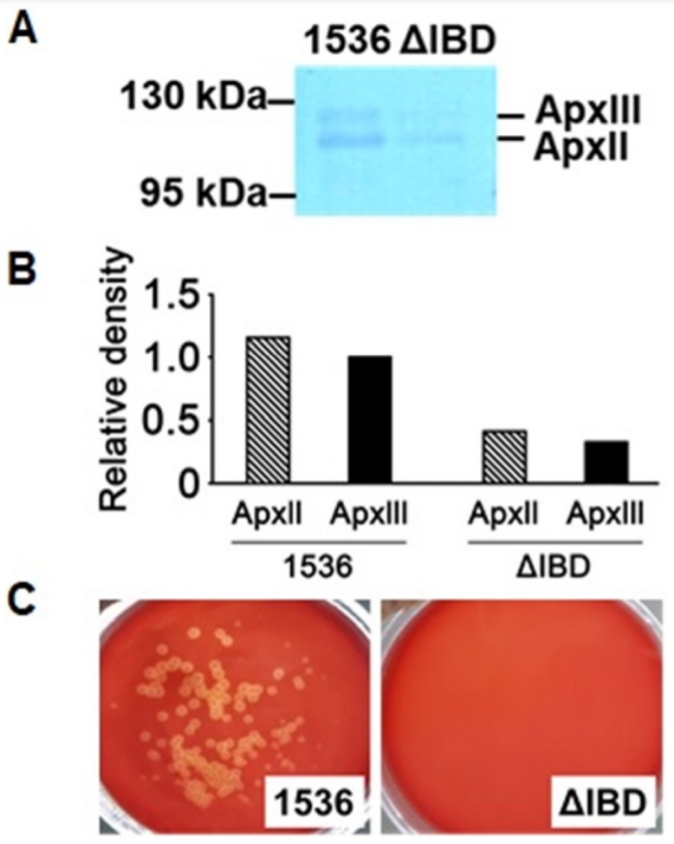
Precipitation of proteins secreted by the App strains into the culture media was performed as described previously [9]. Based upon a western blot analysis using an anti-Apx132 antiserum, the amounts of ApxII and ApxIII proteins released into the culture media by ΔIBD were much lower than those by 1536 (Fig. 3A and 3B). These findings suggest that the inactivation of the apxIB and apxID genes inhibits the secretion of both ApxII and ApxIII.
ApxII is weakly hemolytic, but ApxIII is nonhemolytic [1]. To examine the hemolytic activity of ΔIBD, it was spread on the defibrinated sheep blood agar with 250 μg/mL β-NAD and incubated for 22 h at 37°C. The 1536 strain showed hemolytic activity, as demonstrated by the clear zones surrounding the colonies on the defibrinated sheep blood agar (Fig. 3C). No visible colonies were observed on the ΔIBD agar. It is possible that the growth of the ΔIBD colonies was delayed compared to that of the 1536 colonies and also that the ΔIBD colonies were not viable on the agar.
In summary, we developed a mutant strain of App serotype 2, lacking functional apxIB and apxID genes. The mutant strain exhibited a compromised capacity for the release of ApxII and ApxIII toxins, suggesting that the ApxIB/ApxID exporter is involved in the release of both of the bacterial toxins. Further studies are warranted to assess the relative specificities of the apxIB/apxID transporter to the two toxins.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print